

![]()
![]()
![]()
{maldipickr} provides documented and tested R functions
that will help you dereplicate MALDI-TOF data and cherry-pick
representative spectra of microbial isolates.
{maldipickr} is available on the CRAN and on GitHub.
To install the latest CRAN release, use the following command in R:
install.packages("maldipickr")Or if you are using {renv},
use:
renv::install("maldipickr")To install the development version, use the following command in R:
remotes::install_github("ClavelLab/maldipickr", build_vignettes = TRUE)
# or with renv::install("ClavelLab/maldipickr") Start off with the Introduction to maldipickr for a quickstart. Otherwise, the comprehensive vignettes will walk you through the package functions and showcase how to:
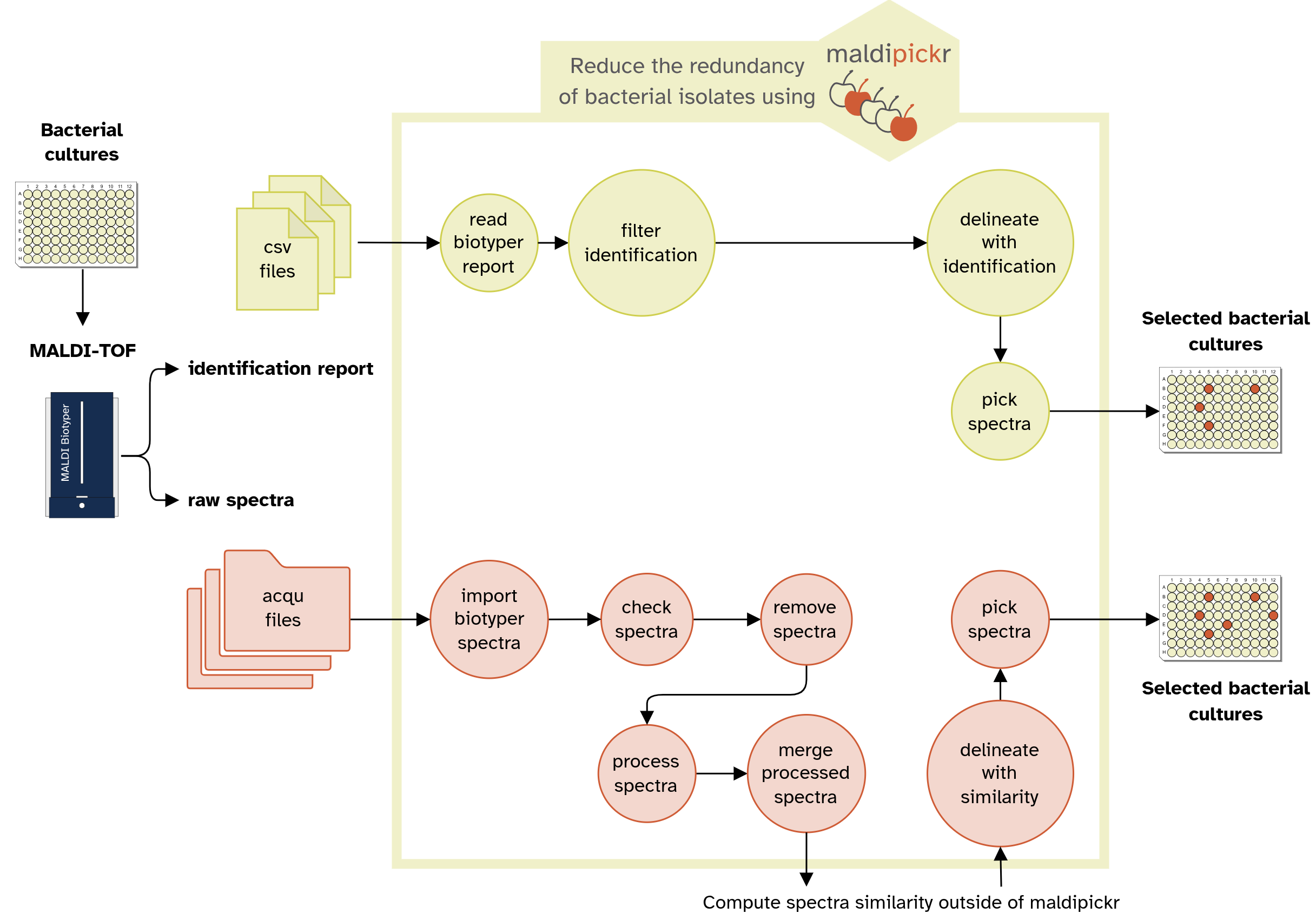
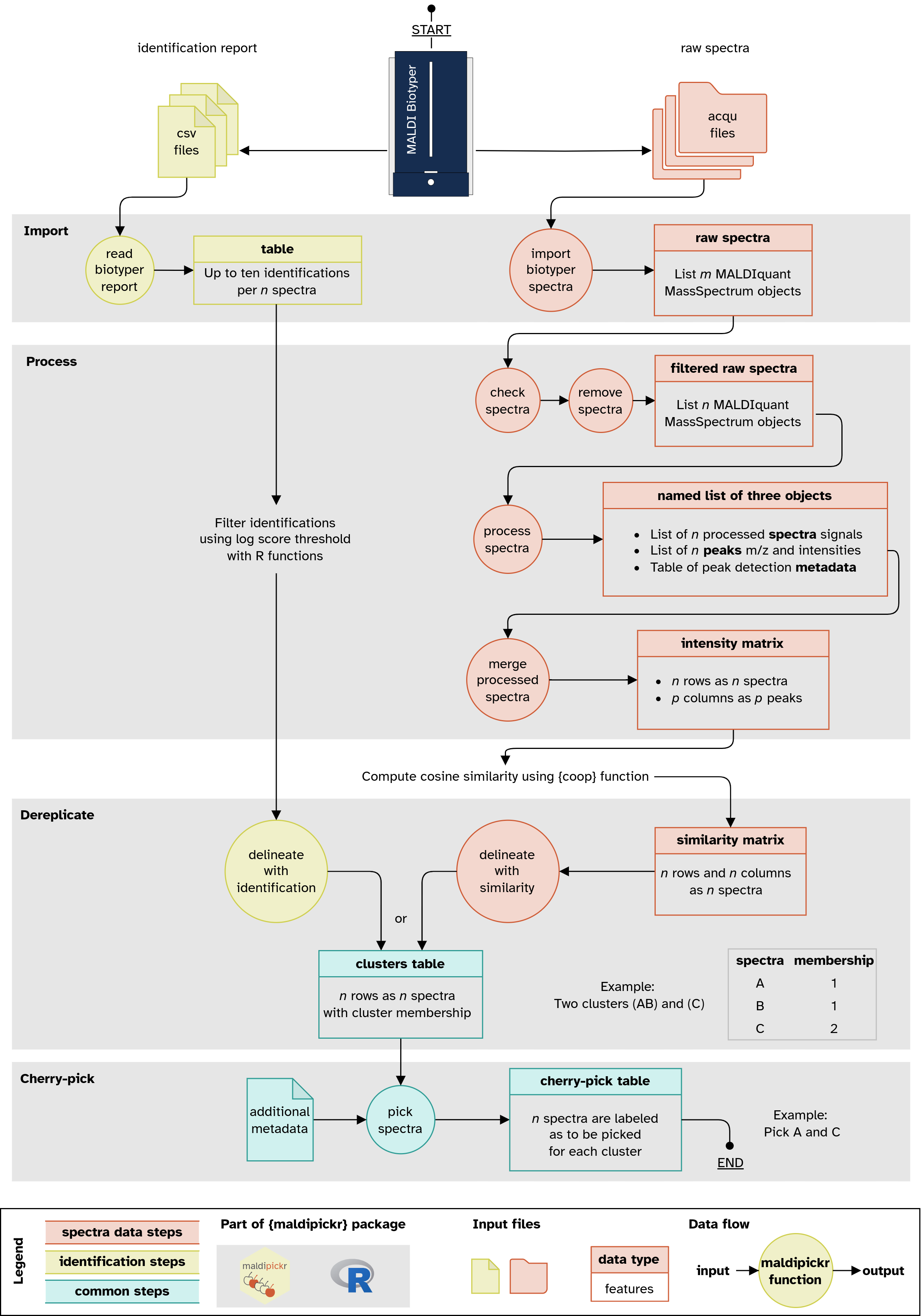
Find a detailed version of the workflow diagram in
man/figures/maldipickr-data-flow-portrait.png
Troubleshoot If something unexpected happened when using this package, please first search the current open or closed issues to look for similar problems. If you are the first, you are more than welcome to open a new issue using the “Bug report” template with a minimal reprex.
Contribute All contributions are welcome and the CONTRIBUTING.md
documents how to participate. Please note that the {maldipickr}
package is released with a Contributor
Code of Conduct. By contributing to this project, you agree to abide
by its terms.
Acknowledgements This R package is developed for
spectra data generated by the Bruker MALDI Biotyper device. The {maldipickr}
package is built from a suite of Rmarkdown files using the {fusen}
package by Rochette S (2023). It relies on:
{MALDIquant}
package from Gibb & Strimmer (2012) for spectra functionsDisclaimer The developers of this package are part of the Clavel Lab and are not affiliated with the company Bruker, therefore this package is independent of the company and is distributed under the GPL-3.0 License. The hexagonal logo was created by Charlie Pauvert and uses the Atkinson Hyperlegible font font and a color palette generated at coolors.co.
If you use our package, please consider citing our work:
Charlie Pauvert, David Wylensek, Selina Nüchtern, Thomas Clavel, maldipickr dereplicates microbial MALDI-TOF spectra to facilitate multiplexed isolation, Bioinformatics Advances, 2026; vbag171, https://doi.org/10.1093/bioadv/vbag171
All the code for the analysis in the manuscript is available at ClavelLab/maldipickr_manuscript or at a CodeOcean capsule (10.24433/CO.1202249.v2) using data at zenodo.org/10.5281/zenodo.15744631.
Matrix-Assisted Laser Desorption/Ionization-Time-Of-Flight (MALDI-TOF)↩︎
{kind=link}