| Type: | Package |
| Title: | 'lme4' for Genomic Selection |
| Version: | 0.1 |
| Date: | 2025-04-06 |
| Maintainer: | Paulino Perez Rodriguez <perpdgo@colpos.mx> |
| Depends: | R (≥ 3.6.3), lme4, methods, Matrix |
| Suggests: | BGLR |
| Description: | Flexible functions that use 'lme4' as computational engine for fitting models used in Genomic Selection (GS). GS is a technology used for genetic improvement, and it has many advantages over phenotype-based selection. There are several statistical models that adequately approach the statistical challenges in GS, such as in linear mixed models (LMMs). The 'lme4' is the standard package for fitting linear and generalized LMMs in the R-package, but its use for genetic analysis is limited because it does not allow the correlation between individuals or groups of individuals to be defined. The 'lme4GS' package is focused on fitting LMMs with covariance structures defined by the user, bandwidth selection, and genomic prediction. The new package is focused on genomic prediction of the models used in GS and can fit LMMs using different variance-covariance matrices. Several examples of GS models are presented using this package as well as the analysis using real data. For more details see Caamal-Pat et.al. (2021) <doi:10.3389/fgene.2021.680569>. |
| License: | GPL-2 | GPL-3 [expanded from: GPL (≥ 2)] |
| NeedsCompilation: | no |
| Packaged: | 2025-04-07 18:10:32 UTC; pperez |
| Author: | Diana Yanira Caamal Pat [aut], Paulino Perez Rodriguez [aut, cre], Erick Javier Suarez Sanchez [ctb] |
| Repository: | CRAN |
| Date/Publication: | 2025-04-08 15:30:02 UTC |
lme4 for Genomic Selection
Description
Flexible functions that use 'lme4' as computational engine for fitting models used in Genomic Selection (GS). GS is a technology used for genetic improvement, and it has many advantages over phenotype-based selection. There are several statistical models that adequately approach the statistical challenges in GS, such as in linear mixed models (LMMs). The 'lme4' is the standard package for fitting linear and generalized LMMs in the R-package, but its use for genetic analysis is limited because it does not allow the correlation between individuals or groups of individuals to be defined. The 'lme4GS' package is focused on fitting LMMs with covariance structures defined by the user, bandwidth selection, and genomic prediction. The new package is focused on genomic prediction of the models used in GS and can fit LMMs using different variance-covariance matrices. Several examples of GS models are presented using this package as well as the analysis using real data. For more details see Caamal-Pat et.al. (2021) <doi:10.3389/fgene.2021.680569>.
Fits a linear mixed model with user specified variance covariance-matrices.
Description
Fits a linear mixed model with user specified variance covariance-matrices.
Usage
lmerUvcov(formula, data = NULL, Uvcov = NULL,verbose=0L)
Arguments
formula |
a two-sided linear formula object describing both the fixed-effects and random-effects part of the model, with the response on the left of a ‘~’ operator and the terms, separated by ‘+’ operators, on the right. Random-effects terms are distinguished by vertical bars (‘|’) separating expressions for design matrices from grouping factors. |
data |
an optional data frame containing the variables named in ‘formula’. |
Uvcov |
list. |
verbose |
integer scalar, verbose output from optimizeLmer function?. If '> 0' verbose output is generated during the optimization of the parameter estimates, default value is 0L. |
Details
The routine fits the linear mixed model:
y=Xβ + Z1 u1 + ... + Zq uq + e,where \boldsymbol y is the response vector, \boldsymbol X is the matrix for
fixed effects, β is the vector of fixed effects,
Zj is a design matrix for random effects,
uj is a vector of random effects,
j=1,\dots,q. We assume that
uj~N(0,σ2j K j),
j=1,\dots,q and
e~N(0,σ2eI).
The linear mixed model can be re-written as:
y=Xβ + Z1* u1*+...+Zq* uq*+e,where Zj*=Zj Lj, with Lj from Cholesky factorization for Kj. Alternatively, Zj*=ZjΓjΛ1/2, with Γj and Λj the matrix of eigen-vectors and eigen-values obtained from the eigen-value decomposition for Kj. The factorization method for Kj is selected automatically at runtime.
Value
An object of class merMod (more specifically,
an object of subclass lmerMod), for which many methods
are available (e.g. methods(class="merMod"))
Author(s)
Paulino Perez-Rodriguez
References
Caamal-Pat D., P. Perez-Rodriguez, J. Crossa, C. Velasco-Cruz, S. Perez-Elizalde, M. Vazquez-Pena. 2021. lme4GS: An R-Package for Genomic Selection. Front. Genet. 12:680569. doi: 10.3389/fgene.2021.680569 doi: 10.3389/fgene.2021.680569
Examples
library(BGLR)
library(lme4GS)
########################################################################
#Example wheat
########################################################################
data(wheat)
X<-wheat.X
Z<-scale(X,center=TRUE,scale=TRUE)
G<-tcrossprod(Z)/ncol(Z)
A<-wheat.A
rownames(G)<-colnames(G)<-rownames(A)
y<-wheat.Y[,1]
data<-data.frame(y=y,m_id=rownames(G),a_id=rownames(A))
fm1<-lmerUvcov(y~(1|m_id)+(1|a_id),data=data,
Uvcov=list(m_id=list(K=G),a_id=list(K=A)))
summary(fm1)
#Predictions
plot(y,predict(fm1))
#Random effects
ranef(fm1)
User defined variance covariance mixed-effects model fits
Description
A mixed-effects model fit by lmerUvcov.
This class extends class "merMod" class and includes one
additional slot, relfac, which is a list of (left) Cholesky
factors of the relationship matrices derived from
user provided variance covariance matrixes.
Objects from the Class
Objects are created by calls to the
lmerUvcov function.
Extends
Class "merMod", directly.
Methods
- ranef
signature(object = "lmerUvcov"): incorporates the user defined variance covariance matrix into the random effects as returned for the object viewed as a"merMod"object.
See Also
Examples
showClass("lmerUvcov")
Genomic relationship matrix for maize lines
Description
A matrix with relationship between individuals for parents of two heterotic groups. The matrix was computed using 511 SNPs using the function A.mat included in rrBLUP package (Endelman, 2011). The row names and column names of this matrix corresponds to the genotype identifiers for Parent 1 and Parent 2.
Phenotypical data for the maize dataset
Description
Phenotypic data for grain yield and plant height for 100 out of 400 possible crosses originated from 40 inbreed lines belonging to two heterotic groups, 20 lines in each. The data are stored in a data.frame with 6 columns: Location, GCA1 (Parent 1), CGA2 (Parent 2), SCA (hybrid), Yield and PlantHeigh. Records with missing values in the last two columns corresponds to hybrids that were not evaluated in the field and that we need to predict.
Extract the conditional means of the random effects
Description
A function to extract the conditional means of the random effects from a fitted model object.
Usage
ranefUvcov(object, postVar = FALSE, drop = FALSE, whichel = names(ans), ...)
Arguments
object |
is an object returned by lmerUvcov. |
postVar |
a logical argument indicating if the conditional variance-covariance matrices of the random effects should be added as an attribute. |
drop |
should components of the return value that would be data frames with a single column, usually a column called “(Intercept)”, be returned as named vectors instead?. |
whichel |
character vector of names of grouping factors for which the random effects should be returned. |
... |
some methods for these generic functions require additional arguments. |
Details
The function ranef extract the conditional means for the liner mixed effects model:
y=Xβ + Z1* u1*+...+Zq* uq*+e,where Zj*=Zj Lj, with Lj from Cholesky factorization for Kj. Alternatively, Zj*=ZjΓjΛ1/2, with Γj and Λj the matrix of eigen-vectors and eigen-values obtained from the eigen-value decomposition for Kj. So, the conditional means of the random effects in the linear mixed effects model:
y=Xβ + Z1 u1 + ... + Zq uq + e,are obtained as follows: ûj=Ljûj* if the Cholesky factorization is used or ûj=ΓjΛj1/2ûj* if the the eigen-value decomposition is used.
Value
A list of data frames, one for each grouping factor.
Author(s)
Paulino Perez-Rodriguez
References
Caamal-Pat D., P. Perez-Rodriguez, J. Crossa, C. Velasco-Cruz, S. Perez-Elizalde, M. Vazquez-Pena. 2021. lme4GS: An R-Package for Genomic Selection. Front. Genet. 12:680569. doi: 10.3389/fgene.2021.680569 doi: 10.3389/fgene.2021.680569
Examples
library(BGLR)
library(lme4GS)
########################################################################
#Example wheat
########################################################################
data(wheat)
X<-wheat.X
Z<-scale(X,center=TRUE,scale=TRUE)
G<-tcrossprod(Z)/ncol(Z)
A<-wheat.A
rownames(G)<-colnames(G)<-rownames(A)
y<-wheat.Y[,1]
data<-data.frame(y=y,m_id=rownames(G),a_id=rownames(A))
fm1<-lmerUvcov(y~(1|m_id)+(1|a_id),data=data,
Uvcov=list(m_id=list(K=G),a_id=list(K=A)))
summary(fm1)
#Predictions
plot(y,predict(fm1))
#Random effects
ranef(fm1)
#Equivalently
ranefUvcov(fm1)
Obtain BLUPs for new levels of random effects with user specified variance covariance-matrices.
Description
Obtain BLUPs for new levels of random effects with user specified variance covariance-matrices.
Usage
ranefUvcovNew(object,Uvcov)
Arguments
object |
is an object returned by lmerUvcov. |
Uvcov |
two level list with ids to be predicted and variance covariance matrix that contains information of these ids and the ids used to fit the model. |
Details
Assume that the random effect uj ~ N(0, σ2jKj) and the matrix Kj is partitioned as follows:
uj=(uj1 uj2)'and
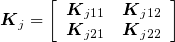
The BLUP for \boldsymbol u_{j2} can be obtained as:
Value
A list of matrix arrays one for each grouping factor
Author(s)
Paulino Perez-Rodriguez
References
Caamal-Pat D., P. Perez-Rodriguez, J. Crossa, C. Velasco-Cruz, S. Perez-Elizalde, M. Vazquez-Pena. 2021. lme4GS: An R-Package for Genomic Selection. Front. Genet. 12:680569. doi: 10.3389/fgene.2021.680569 doi: 10.3389/fgene.2021.680569
Examples
library(BGLR)
library(lme4GS)
data(wheat)
X<-wheat.X
Z<-scale(X,center=TRUE,scale=TRUE)
G<-tcrossprod(Z)/ncol(Z)
A<-wheat.A
rownames(G)<-colnames(G)<-rownames(A)
y<-wheat.Y[,1]
#Predict 10/100 of records selected at random.
#The data were partitioned in 10 groups at random
#and we predict individuals in group 2.
fold<-2
y_trn<-y[wheat.sets!=fold]
y_tst<-y[wheat.sets==fold]
A_trn=A[wheat.sets!=fold,wheat.sets!=fold]
G_trn=G[wheat.sets!=fold,wheat.sets!=fold]
pheno_trn=data.frame(y_trn=y_trn,m_id=rownames(A_trn),a_id=rownames(G_trn))
#######################################################################################
#Marker based prediction
#######################################################################################
fm1<-lmerUvcov(y_trn~1+(1|m_id),data=pheno_trn,Uvcov=list(m_id=list(K=G_trn)))
plot(pheno_trn$y_trn,predict(fm1),xlab="Phenotype",ylab="Pred. Gen. Value")
#BLUP for individuals in the testing set
blup_tst<-ranefUvcovNew(fm1,Uvcov=list(m_id=list(K=G)))
blup_tst<-blup_tst$m_id[,1]
#Comparison
#Check the names
names(y_tst)<-rownames(G)[wheat.sets==fold]
blup_tst<-blup_tst[match(names(y_tst),names(blup_tst))]
yHat_tst<-fixef(fm1)[1]+blup_tst
points(y_tst,yHat_tst,col="red",pch=19)
#Correlation in testing set
cor(y_tst,yHat_tst)
#######################################################################################
#Pedigree based prediction
#######################################################################################
fm2<-lmerUvcov(y_trn~1+(1|a_id),data=pheno_trn,Uvcov=list(a_id=list(K=A_trn)))
plot(pheno_trn$y_trn,predict(fm2),xlab="Phenotype",ylab="Pred. Gen. Value")
#BLUP for individuals in the testing set
blup_tst<-ranefUvcovNew(fm2,Uvcov=list(a_id=list(K=A)))
blup_tst<-blup_tst$a_id[,1]
#Comparison
#Check the names
names(y_tst)<-rownames(A)[wheat.sets==fold]
blup_tst<-blup_tst[match(names(y_tst),names(blup_tst))]
yHat_tst<-fixef(fm2)[1]+blup_tst
points(y_tst,yHat_tst,col="red",pch=19)
#Correlation in testing set
cor(y_tst,yHat_tst)
#######################################################################################
#Markers + Pedigree based prediction
fm3<-lmerUvcov(y_trn~1+(1|m_id)+(1|a_id),data=pheno_trn,
Uvcov=list(m_id=list(K=G_trn),a_id=list(K=A_trn)))
plot(pheno_trn$y_trn,predict(fm3),xlab="Phenotype",ylab="Pred. Gen. Value")
#BLUP for individuals in the testing set
blup_tst<-ranefUvcovNew(fm3,Uvcov=list(m_id=list(K=G),a_id=list(K=A)))
blup_tst_m<-blup_tst$m_id[,1]
blup_tst_a<-blup_tst$a_id[,1]
#Comparison
#Check the names
names(y_tst)<-rownames(A)[wheat.sets==fold]
blup_tst_m<-blup_tst_m[match(names(y_tst),names(blup_tst_m))]
blup_tst_a<-blup_tst_a[match(names(y_tst),names(blup_tst_a))]
yHat_tst<-fixef(fm3)[1] + blup_tst_m + blup_tst_a
points(y_tst,yHat_tst,col="red",pch=19)
#Correlation in testing set
cor(y_tst,yHat_tst)
Selection of bandwidth parameter for Gaussian and exponential kernels.
Description
Obtain the optimal value of the bandwidth parameter for the Gaussian and exponential kernels.
Usage
theta_optim(formula, data = NULL, Uvcov = NULL,
kernel = list(D = NULL, kernel_type = "gaussian",
theta_seq = NULL, MRK = NULL),
verbose_lmer= 0L, verbose_grid_search=0L)
Arguments
formula |
a two-sided linear formula object describing both the fixed-effects and random-effects part of the model, with the response on the left of a ‘~’ operator and the terms, separated by ‘+’ operators, on the right. Random-effects terms are distinguished by vertical bars (‘|’) separating expressions for design matrices from grouping factors. |
data |
an optional data frame containing the variables named in ‘formula’. |
Uvcov |
list. |
kernel |
list with the following elements, i)D: Distance matrix (can be NULL), ii) kernel_type: character, can be either "gaussian" or "exponential", ii)theta_seq: sequence of values for theta from which we select the optimum (can be NULL), iv) MRK: marker matrix from wich Euclidean distance is computed (can be NULL). |
verbose_lmer |
integer scalar, verbose output from optimizeLmer function?. If '> 0' verbose output is generated during the optimization of the parameter estimates, default value is 0L. |
verbose_grid_search |
integer scalar, if '>0' verbose output is generated, default value is 0L. |
Value
A list that contains:
LL |
Log-likelihood. |
LL.max |
Maximum of likelihood. |
theta |
Sequence of values for the bandwidth. |
theta.max |
Value of bandwidth when log-likelihood attains the maximum. |
fm |
Fitted model with the optimum bandwidth parameter. |
K.opt |
The kernel for the optimum bandwith parameter. |
Author(s)
Paulino Perez-Rodriguez, Diana Caamal-Pat
References
Caamal-Pat D., P. Perez-Rodriguez, J. Crossa, C. Velasco-Cruz, S. Perez-Elizalde, M. Vazquez-Pena. 2021. lme4GS: An R-Package for Genomic Selection. Front. Genet. 12:680569. doi: 10.3389/fgene.2021.680569 doi: 10.3389/fgene.2021.680569
Examples
library(BGLR)
library(lme4GS)
data(wheat)
y = wheat.Y[,1]
X = wheat.X
A = wheat.A
rownames(X) <- rownames(A)
#model y=1*mu+Z_1*u_1+e, u_1~NM(0, \sigma_1*KG), KG: Gaussian kernel
wheat = data.frame(y=y, k_id=rownames(X))
fm1 <- theta_optim(y~(1|k_id), data = wheat, Uvcov = NULL,
kernel = list(D = NULL, kernel_type = "gaussian",
theta_seq = seq(3,8,length.out=10), MRK = X),
verbose_lmer=0L,verbose_grid_search=1L)
plot(fm1$theta,fm1$LL,xlab=expression(theta),ylab="Log-Likelihood")
fm1$theta.max
fm1$LL.max
Pedigree infomation for 599 wheat lines
Description
A data.frame with 3 columns: gpid1 and gpid2 which corresponds to the GID of parents 1 and 2 respectively, progenie which corresponds to the GIDs of progenie.
Phenotypical data for the wheat dataset
Description
A data.frame with 4 columns, Env for environments, Rep for replicates, GID for genotype identifiers, Yield for grain yield.
Molecular markers
Description
Is a matrix (599 x 1279) with DArT genotypes; data are
from pure lines and genotypes were coded as 0/1 denoting
the absence/presence of the DArT. Markers with a
minor allele frequency lower than 0.05 were removed, and
missing genotypes were imputed with samples from the
marginal distribution of marker genotypes, that is, x_{ij}=Bernoulli(\hat p_j), where \hat p_j
is the estimated allele frequency computed from the non-missing genotypes. The number of DArT
MMs after edition was 1279.
Source
International Maize and Wheat Improvement Center (CIMMYT), Mexico.
Sets for cross validation (CV)
Description
Is a vector (599 x 1) that assigns observations to 10 disjoint sets; the assignment was generated at random. This is used later to conduct a 10-fold CV.
Source
International Maize and Wheat Improvement Center (CIMMYT), Mexico.